



|
Antigenic evolution of viruses in host populations
Par Igor Rouzine (Sorbonne Université - LCQB)
Le 15 Janvier 2019 à 11h00 - Salle de séminaires 5ème étage, Tour 32-33
|
Résumé
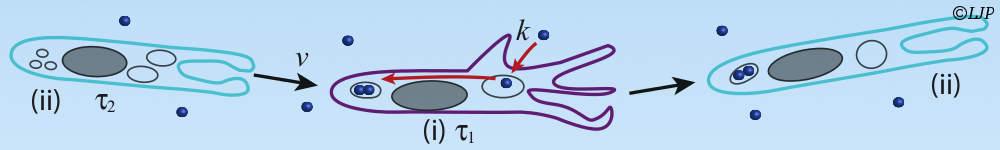
To escape immune recognition in previously infected hosts, viruses evolve genetically in immunologically important regions. The host’s immune system responds by generating new memory cells recognizing the mutated viral strains. Despite recent advances in data collec- tion and analysis, it remains conceptually unclear how epidemiology, immune response, and evolutionary factors interact to produce the observed speed of evolution and the inci- dence of infection. Here we establish a general and simple relationship between long-term cross-immunity, genetic diversity, speed of evolution, and incidence. We develop an analytic method fusing the standard epidemiological susceptible-infected-recovered approach and the modern virus evolution theory. The model includes the factors of strain selection due to immune memory cells, random genetic drift, and clonal interference effects. We predict that the distribution of recovered individuals in memory serotypes creates a moving fitness land- scape for the circulating strains which drives antigenic escape. The fitness slope (effective selection coefficient) is proportional to the reproductive number in the absence of immunity R0 and inversely proportional to the cross-immunity distance a, defined as the genetic dis- tance of a virus strain from a previously infecting strain conferring 50% decrease in infection probability. Analysis predicts that the evolution rate increases linearly with the fitness slope and logarithmically with the genomic mutation rate and the host population size. Fitting our analytic model to data obtained for influenza A H3N2 and H1N1, we predict the annual infection incidence within a previously estimated range, (4-7)%, and the antigenic mutation rate of Ub = (5 − 8) 10−4 per transmission event per genome. Our prediction of the cross- immunity distance of a = (14 − 15) aminoacid substitutions agrees with independent data for equine influenza.